Dehydratace je nedostatek tekutin v těle. Ohrožuje především děti a staré lidi.

Dehydratace u dětí
Dehydratace je nedostatek tekutin v těle. Ohrožuje především děti a staré lidi.
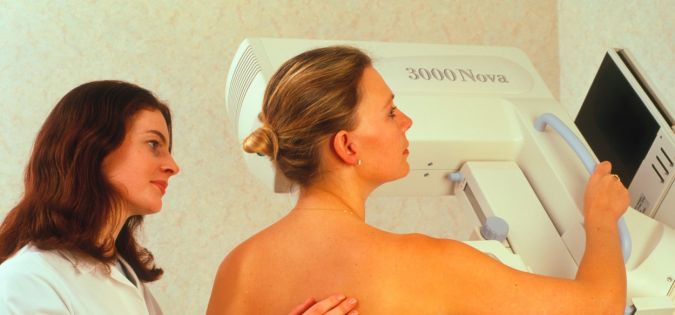
Mamografie je rentgenové vyšetření. Vyšetření podstupují ženy preventivně nebo při podezření na onemocnění prsou.
Vředová choroba je onemocnění, které postihuje mnoho lidí v naší populaci. Vředy bývají nejčastěji ve sliznici jícnu, žaludku anebo přilehlé části tenkého střeva, dvanáctníku. Jedná se o defekty sliznice, které se projevují různými zažívacími problémy, často i bolestí.
Menstruační cyklus je pravidelně se opakující proces ženského těla, při kterém dochází na základě hormonálních změn k odlučování děložní sliznice. Menstruace se u žen pravidelně opakuje od puberty a ustává až během menopauzy.
Bolest je nepříjemný pocit, zkušenost sestávající z odpovědi na škodlivý podnět. Je to obranný mechanismus organismu před poškozením či zraněním. Bolest vzniká drážděním nervových zakončení v kůži a v orgánech, které pomocí nervových vláken vysílají vzruch do míchy a vyšších struktur v mozku.
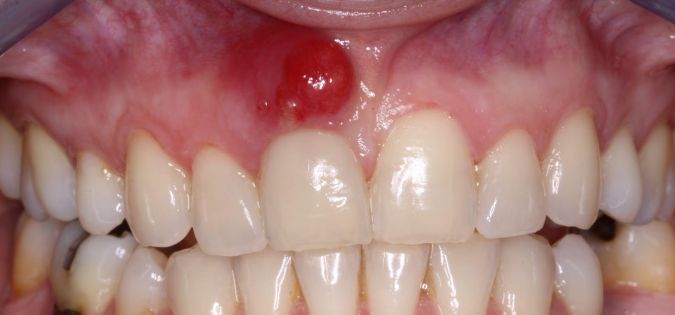
Abscesy neboli hlízy jsou ohraničená zánětlivá ložiska, která se mohou tvořit kdekoliv uvnitř těla nebo na jeho povrchu. Jejich rozměry se různí od drobných puchýřků a vřídků na kůži nebo při vlasových koříncích až po závažnější abscesy zubních lůžek nebo abscesy vnitřních orgánů, například při zánětu slepého střeva (apendixu).
Zvýšený cholesterol v krvi je rizikovým faktorem pro rozvoj aterosklerózy cév, což následně vede ke kardiovaskulárním onemocněním, jako jsou srdeční infarkt, cévní mozková příhoda, plicní embolie anebo ischemická choroba dolních končetin. Velmi významnou roli v léčbě hraje správná životospráva.
Nedaří se vám zhubnout? Trpíte často zácpou a potřebujete si upravit zažívání? Na tyto a mnohé další problémy existuje osvědčená rada - zvyšte příjem vlákniny. Poradíme vám, v jakých potravinách jí je nejvíce.
Homocystein je aminokyselina, která vzniká v těle při metabolismu jiné aminokyseliny – methioninu. Změny v koncetraci homocysteinu jsou dávány do souvislosti s cukrovkou, kardiovaskulárními chorobami, demencí atd. Poruchy hladiny homocysteinu v krvi jsou dány jednak geneticky, jednak naší stravou.
Množství kožních nádorů v poslední době stoupá. Jedním z nejobávanějších kožních nádorů je jistě melanom. Za rizikový faktor se považuje především nadměrné vystavování kůže UV záření. Ochrana před sluncem by měla být tedy samozřejmostí.
Hysteroskopie je šetrná metoda, která se používá jak za diagnostickým účelem, tak za účelem operačním.
Pandemie COVID-19 ovlivňuje globálně nejenom fyzické zdraví, ale také to psychické. Již nyní můžeme s jistotu říci, že v posledním roce přibylo pacientů s úzkostnými poruchami, depresí, post-traumatickým stresem a dalšími stresovými poruchami.
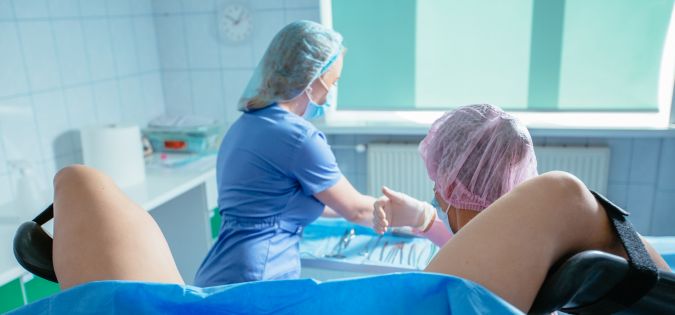
Císařský řez je způsob porodu, při kterém se řezem protne břišní stěna a děloha matky. Důvodem rozhodnutí lékaře pro císařský řez mohou být například malé rozměry pánve, příliš velký plod nebo jeho nevhodná poloha (boční, pánevním koncem apod.) Někdy je důvodem pro císařský řez příliš nízko uložená placenta, která by normálnímu porodu buď zcela bránila, nebo by mohla vyvolat masivní krvácení.
Porod a poporodní období je čas příchodu očekávaného potomka do rodiny a první chvíle s ním strávené. Zbývá jen rozhodnout o přítomnosti otce u porodu a jméně dítěte. Porod je rozdělen do tří fází, které začínají pravidelnými kontrakcemi, pokračuje usilovným tlačením k vypuzení plodu a končí porodem dítěte a placenty. Dítěti se podvazuje a přestřihuje pupečník, je osušen, vyšetřen a zabalen. Žena po porodu odpočívá a čeká na tvorbu mléka ke kojení, zpravidla opouští porodnici 4. nebo 5.den a pak nastává doba šestinedělí.
Nezdravá výživa v těhotenství mívá na vývoj plodu neblahý vliv. Mezi nevhodné potraviny a nápoje patří například kofein, alkohol a jiné povzbudivé látky jako třeba chinin, dále uzeniny, nadměrně kořeněná a slaná jídla, jídla typu fast food a nadměrné množství cukrovinek. Neblahý vliv na plod má také kouření, užívání drog a taktéž vegetariánství a veganství.
Očkování je v současné době (2022) doporučováno všem osobám starším 12 let, včetně těhotných a kojících žen. Podle nejrůznějších studií benefity očkování jasně převyšují možná rizika. Na internetových fórech koluje velká řada mýtů o očkování, která mohou ztěžovat rozhodování nechat se očkovat. Jak to tedy doopravdy je?
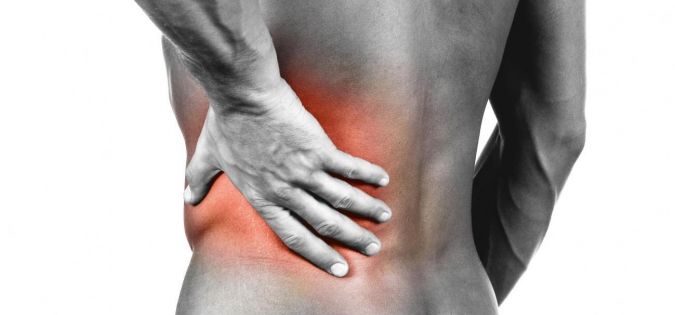
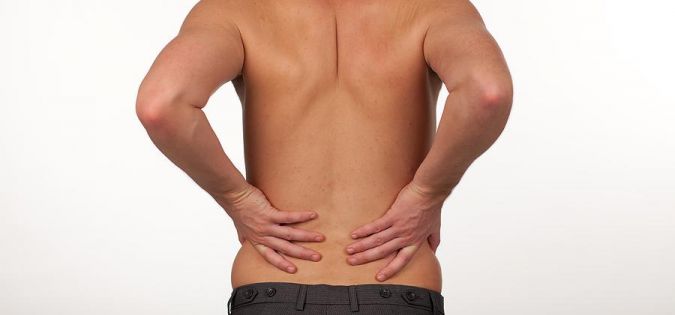
Zatím žádný komentářBolest v kříži je v naší populaci velmi častá obtíž, ve valné většině však způsobená pouze špatnými pohybovými návyky. Bolest v kříži je nejlépe řešit udržením si zdravého životního stylu a zdravých pohybových aktivit. V tom nám může často pomoci i vhodné nastavení židle popřípadě kvalitní matrace a polštář zajišťující pohodlný a klidný spánek.
35 komentářeDobrý den, mohu se optat jdu na gastroskopii a mám veliký strach, nikdy sem na tomto ...
29.ledna letošního roku jsem byl operován na TEP kyčle. Po 6ti týdnech už jsem chodil...